Motivations
- Why do some genomes evolve fast, some slow?
- Does abundance determine genetic polymorphism levels? Selection efficiency?
- What is the proportion of adaptive amino-acid substitutions?
- Why do some species, but not all, optimize codon usage?
- What controls base composition variations within and between genomes?
- Why is the mitochondrial genome hypermutable in animals, but not in plants?
- Does self-fertilization impede molecular adaptation?
These and other basic questions regarding molecular evolution are not answered yet, or only vaguely, despite the huge amount of data available, and the strength of the population genetic theory. One limiting factor is probably the taxonomic bias of current population genomic data sets, especially in animals - most of the evolutionary genomic literature is in mammals or Drosophila. The PopPhyl project aims at characterizing within- and between-species molecular variations in a substantial number of metazoan taxa thanks to next-generation sequencing technology, with the hope of linking genome evolutionary patterns to species biology and ecology.
Goals
Making population genetics comparative
We want to approach the neutral (synonymous) and selected (non-synonymous) variation within species (ΠS, ΠN, allele frequency spectra), and between closely-related species (dN, dS), for large numbers of genes, in >30 animal taxa. Such data are indicative of the evolutionary forces at work – mutation biases, distribution of selection coefficients, recombination, genetic drift, GC-biased gene conversion. We will measure these parameters in a diversified panel of species, thus providing a broader view of population genomic processes across animals. We will estimate the ratio of adaptive to neutral amino-acid changes in many species, assess the prevalence of GC-biased gene conversion and codon usage optimization in various phyla, and identify the causes of high substitution rate in fast-evolving lineages.
Linking genome evolution to species biology
Molecular evolution is governed by population genetic parameters (mutation, selection, drift, recombination), which in turn presumably depend on species life-history and ecology. We want to check whether genomic variation patterns reflect species biology according to the population genetic theory. Here are examples of the predictions we want to test:
| Trait | population genetic parameter | predicted genomic pattern |
|---|---|---|
| marine (vs terrestrial) small (vs large) |
large population size | high within-species diversity low ΠN/ΠS ratio |
| selfer (vs outcrosser) | low recombination rate small population size |
high ΠN/ΠS ratio slow adaptive rate high homozygosity no biased gene conversion |
| long (vs short) lifespan | low per year mutation rate, selection for somatic repair | low dS (per million year) slow mitochondrial evolution |
Exploring the molecular diversity of non-model animals
Most of the sequencing effort in animals so far has been concentrated in a small number of model laboratory organisms - vertebrates, Drosophila, Caenorhabditis. The PopPhyl project will explore the molecular diversity of neglected taxa (molluscs, annelids, nemertians, cnidarians), emblematic animals (Galapagos tortoise, king penguin, bath sponge), and species showing remarkable life history or ecology (hydrothermal annelids, 400-year old bivalves, social insects), with the hope of discovering new genes and adaptations, and providing information relevant to conservation biology and environmental sciences.
Approach
Field work
More than 30 focal species spanning the metazoan diversity have been selected based on their biology and ecology. For each of them, 10 living individuals are sampled across the species geographic range with the help of expert colleagues . Two closely-related outgroup species are identified, and sampled as well (one or two individuals each). See pictures here.
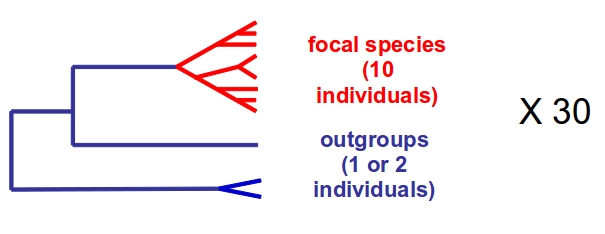
PopPhyl sampling scheme
Transcriptome data acquisition
Total RNA will be extracted from the ~500 individuals sampled in PopPhyl using standard and modified protocols. Then individual transcriptomes will be sequenced using a combination of next-generation technologies. For each focal group, one ½ 454 run (~500,000 reads of length ~400) will be performed from total RNA of one or several pooled individuals. Such long reads are useful for proper assembly of cDNA contigs. In addition, individual transcriptomes will be separately sequenced using the Illumina technology (~4,000,000 reads of length ~100 per individual). SNPs and genotypes will be deduced from the mapping of short reads on assembled reference transcriptomes.
Bioinformatics
The PopPhyl data analysis pipeline should look like:
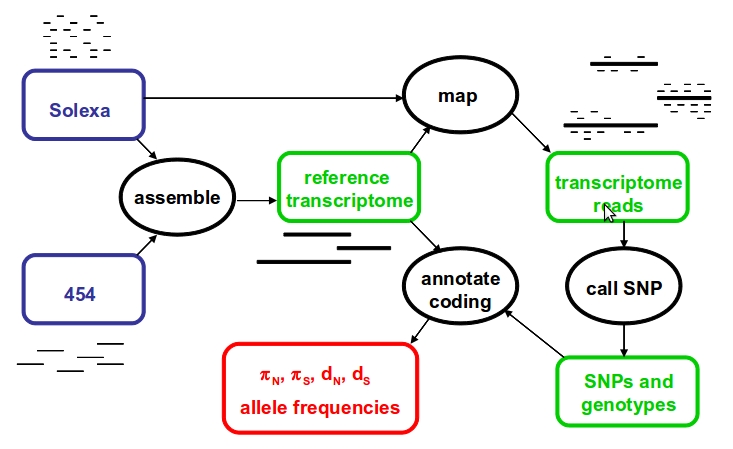
Transcriptome assembly, short read mapping and coding sequence annotation will be achieved using publicly available software, encapsulated in appropriate C++/perl/bash code. SNP and genotype calling algorithms will be developed in house. Methods will run on a dedicated cluster hosted by the Montpellier Bioinformatics & Biodiversity platform.
Funded by: European Research Council
Hosted by: CNRS – UMR 5554 – Institut des Sciences de l'Evolution